Molecular genetics of polyposis and hereditary colorectal cancer
Paolo Radice1, Alessandro Cama2, Renato Mariani-Costantini2
1 Divisione di Oncologia Sperimentale A,
Istituto Nazionale Tumori, Milano;
2 Cattedra di Patologia Generale, Università "Gabriele
D'Annunzio", Chieti, Italy;
Key words: FAP, HNPCC, APC, mismatch repair
Running title:
Although the genetics of inherited predisposition to colorectal carcinomas is still far to be completely elucidated, significant acquisitions have been achieved in the last few years. In fact, the genetic determinants of two major syndromes associated with susceptibility to colorectal cancer, Familial Adenomatous Polyposis (FAP) and Hereditary Non Polyposis Colorectal Cancer (HNPCC) have been identified. FAP appears to be a monogenic disease, due to mutations in the APC gene. This gene encodes a large protein, that appears to be involved in the control of signal transmission from the extracellular environment to the cytoskeleton and vice versa. Most APC gene mutations result in truncated APC proteins. The site of the germline APC mutations correlates with phenotypic manifestations of the disease. A number of studies demonstrated that, accordingly with Knudson's hypothesis, biallelic inactivation of the APC gene occurs in both colorectal and extracolorectal tumours associated with FAP. In contrast with FAP, HNPCC is characterised by a high degree of genetic heterogeneity and four different genes, namely hMSH2, hMLH1, hPMS1 and hPMS2 have been found to be mutated in affected individuals. These genes, termed DNA mismatch repair (MMR) genes, are the human counterparts of loci that in lower organisms control the fidelity of DNA replication. The discovery of human MMR genes has led to the identification of a new mechanism of carcinogenesis based on the acquisition by the cell of a so called "mutator phenotype".
Colorectal cancer is one of the most common forms of malignancy, ranking in second to third position with regard to overall frequency in the developed countries of the Western hemisphere (1). "Sporadic" forms are thought to account for about 90% of all colon cancers occurring each year, even though the term "sporadic" should be interpreted with caution, since first degree relatives of colorectal cancer patients have a 2-3-fold increased risk for colon cancer. Such excess risk could be at least partly related to genetic components (2,3). Well defined forms of hereditary colorectal cancer are currently thought to account for about 10% of the total number of colorectal cancers (4). Hereditary polyposis syndromes, including familial adenomatous polyposis (FAP) and Peutz-Jeghers syndrome, account for a subset of cases estimated at about 1% of the total number of colorectal cancers, whereas hereditary non-polyposis colorectal cancer, (HNPCC, also known as Lynch syndrome) is considered responsible for the remaining 5-10% of the cases (4). FAP and HNPCC are highly penetrant, are associated with colorectal cancer development at young or relatively young age and are of crucial scientific and social importance. The last few years witnessed very exciting advances in the identification of genes responsible for FAP and HNPCC and the present review is focused on the genetics of these two important forms of hereditary cancer.
Familial Adenomatous Polyposis (FAP)
FAP is a hereditary dominant autosomal disease with complete penetrance, that has estimated population frequencies ranging from 1:5000 - 1:8000 in Western countries to 1:17000 in Japan (5). In its classic presentation, the disease is typically characterised by the development of hundreds to thousands adenomatous polyps of the colorectum, that are usually manifest by the second or third decade of life (4,5). In the absence of follow-up and surgical treatment, adenocarcinomas invariably develop by the fourth decade of life (6). FAP is also associated with a variety of extracolorectal manifestations, that include congenital hypertrophy of the retinal pigment epithelium (CHRPE), gastro-duodenal and jejunal-ileal polyps, dental anomalies, cranial and mandibular osteomas and exostoses, epidermoid cysts, subcutaneous fibromas, and locally aggressive abdominal and retroperitoneal desmoids (4,5). The presence of epidermoid cysts, subcutaneous fibromas, osteomas, dental anomalies and desmoid tumours characterise a variant of FAP that is designated Gardner's syndrome (4). Desmoids are particularly important among extracolorectal FAP-associated neoplasms, because these invasive proliferations of fibroblasts represent one of the main causes of death in surgically-treated FAP patients (4-7). In addition to the classic variant of FAP and to Gardner's syndrome, recent clinical and genetic studies contributed to the definition of an attenuated FAP variant, designated attenuated adenomatous polyposis coli (AAPC), characterised by a relatively delayed onset and by the presence of a reduced number of colorectal polyps, generally well below 100 (8-10). The early identification of FAP carriers is of the utmost importance, since colorectal cancer can be efficiently prevented if the patient is correctly followed and undergoes an appropriately timed colectomy. In addition, medical therapy with non-steroid anti-inflammatory drugs, such as sulindac, may help to control the development of colorectal adenomas in disease carriers (5-6). The detection of early clinical markers of the disease, that may be present from birth, such as CHRPE (4-6), facilitates the timely diagnosis of FAP in at risk subjects. However, depending on the samples of FAP patients studied, CHRPE occurs with a frequency ranging from 66% to 97% of the cases and may also occur, in milder form, in unaffected subjects. Moreover, there are significant differences in the genetic predisposition to CHRPE in different FAP kindreds (11). Fortunately, following the identification of the gene responsible for FAP, designated adenomatous polyposis coli (APC) gene, and the description of germline mutations responsible for the disease (12-15), it is now possible to diagnose the disease-carrier status at the genetic level in a high percentage of cases. In this respect, FAP represents a model for the rapid transfer of molecular diagnosis to clinical practice.
Structure and functions of the APC gene
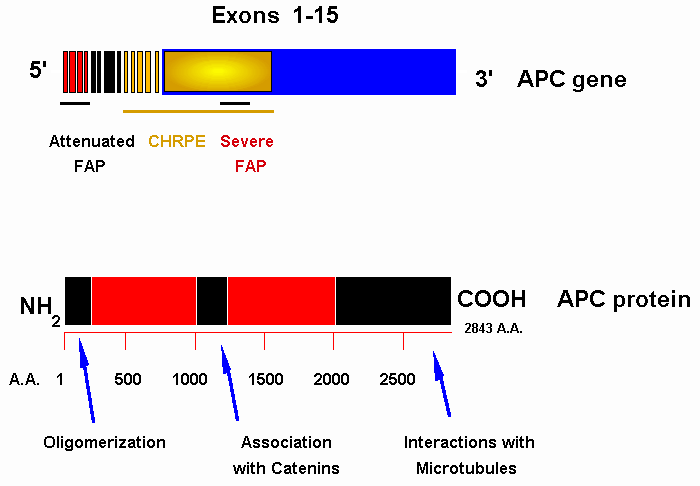
The locus responsible for FAP was localised in 1986, after cytogenetic and linkage studies, and mapped to 5q21-22, a region that frequently shows losses of heterozygosity in colorectal cancer (16-18). The APC gene was isolated in 1991 (12-15). Its coding region spans at least 8538 bp (i.e., 2843 codons), and is organised in 15 exons, the last of which containing more than 6000 nucleotides (12,14). At least 4 additional exons 5' to exon 1 were also identified (19,20). These exons are transcribed in various splicing combinations in a number of tissues, including brain, where a specific transcript, containing an additional exon upstream of exon 1, has been consistently identified (19). Moreover, an inter-gene transcript is generated by alternative splicing between exon 14 of the APC gene and SRP19, the gene encoding the 19 kDa signal recognition particle protein (13,14, 19). At present, there is still no firm evidence concerning the expression and possible tissue specificities of protein products from differently spliced forms of the APC gene. In any case, the presence of different inter- and intra-gene APC splicing forms suggests that the APC gene has multiple functions. Several putative promoter elements have been identified in the region upstream of APC. Further studies are needed to clarify the significance of elements existing in this region in the determination of transcription initiation sites and tissue-specific forms of expression. The APC gene encodes a 311.8 kDa cytoplasmic protein, that has limited homologies to intermediate filament structural proteins, such as myosins and keratins. Homologies also exist with another gene assigned to 5q21-22 and associated with sporadic colorectal cancer, designated MCC for Mutated in Colon Cancer (12,14). Moreover, the APC gene has a short region of homology with the m3 muscarinic acetylcholine receptor gene, which acts as a regulator of G protein coupling (14,15). In their turn, G proteins are known to regulate the activity of phospholipase A2, an enzyme that generates arachidonic acid, the substrate for prostaglandin synthetase, from phospholipids. In this respect, it is intriguing that sulindac, a cyclooxygenase inhibitor that interferes with prostaglandin synthetase activity, may delay the onset of polyps in FAP patients. The APC protein is capable of forming homodimers. Peptides limited to the first 171 aminoacid residues are still capable of interacting with wild type APC proteins (21). Such function is related to a structural domain formed by the first 45 amino acids, that are necessary for the interaction. The APC protein has been isolated in association with catenins, cytosolic proteins that regulate the functions of cadherins, a family of transmembrane glycoproteins that mediate Ca++ dependent cell-cell adhesion and play important roles in development and differentiation (22, 23). A b-catenin binding domain was identified in the central region of the APC protein, between the amino acid residues 1014 and 1210 (22). This binding site consists in a triple repeated consensus sequence of 15 amino acids. Other b-catenin binding and regulatory sites are located between amino acids 1342 and 2075. By downregulating and redistributing b-catenin, the APC protein appears to exert a key role in the control of cytoplasmic catenin levels (24,25). The carboxy-terminal region of the APC protein contains a domain that binds cytoplasmic microtubules and promotes their assembly (26-27). Thus, based on available data, the APC protein appears to control signal transmission from the extracellular environment to the cytoskeleton and vice versa.
Immunohistochemical studies of the APC gene product in normal colonic epithelium indicate that the protein is concentrated at the basolateral portion of crypt epithelial cells (28). APC immunostaining gradually increases as cells progress from the base of the crypt towards the luminal surface, which suggests an association between APC expression and cell differentiation.
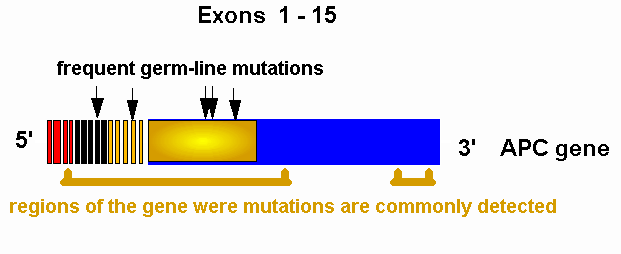
Depending on the methods of mutational analysis used, germline mutations of the APC gene have been found in percentages variable from 60% to over 85% of FAP patients analysed in different studies (29-34). The vast majority of APC mutations reported to date (about 97%) consist in small deletions or insertions or point mutations that introduce premature protein termination signals (35). A relatively small number of missense mutations has also been reported, but, until functional assays will be available for the APC protein, the pathogenic role of such mutations can only be inferred on the basis of their cosegregation with disease in the kindred. The presence of a small subset of FAP patients in which mutations have not been identified is probably related to the limits of the presently used mutation screening technology, which, being based on PCR amplification of the coding region, could conceivably miss some large deletions and all mutations occurring in introns or in upstream or downstream regulatory regions of the gene. For diagnostic purposes, it is relevant to note that germline APC mutations occur mostly in the 5' half of the coding sequence, with two hot spots at codons 1061 and 1309. Mutations at these two codons account together for about 30% of the cases of FAP (29,34).
Several studies analysed correlations between the clinical expression of FAP and the germline APC mutation site. The available data suggest that knowledge of the type and site of APC mutations could contribute to the follow-up of FAP patients and to the evaluation of therapeutic regimens.
Germline mutations occurring within the first 4 coding exons of the gene are correlated with smaller number of polyps, late disease onset and slower progression, characteristics that are observed in AAPC (36). In particular, it has been proposed that the functional boundary separating mutations associated with attenuated phenotype from those determining the classic FAP presentation is located around codons 157-168 of the APC gene. It has been speculated that such mutations result in the expression of unstable mRNAs, or yield very short truncated peptides, that would not efficiently dimerise with wild type products. However, it should be kept in mind that not all APC mutations responsible for AAPC occur upstream of the boundary, as mutations that do not significantly alter the structure of the gene, or that are quantitatively attenuated because of differential splicing, could also result in an attenuated form of FAP (Cama et al., manuscript in preparation).
As determined by the number of adenomas, the highest levels of colorectal disease expression appear to be associated with mutations between codons 1250 and 1464 of the APC gene (37). In particular, a highly penetrant form of FAP, characterised by early onset, often within the first decade of life, and rapid progression appears to be associated with the 5 bp deletion at codon 1309, that represents one of the most common APC mutations (34,38). It has been speculated that this mutation might generate a protein product acting more efficiently as dominant-negative because of a marked ability to interact with and sequester products of the normal allele. Mutations downstream of codon 1445 up to codon 1578 tend to be more frequently associated with extracolorectal disease, including desmoid tumours, osteomas, epidermoid cysts and upper gastrointestinal polyps (39). In particular, mutations downstream of codon 1464 appear to be associated with increased risk of desmoids. The presence of CHRPE, widely used as an early clinical indicator of FAP, is also dependent upon the location of the germline mutation in the APC gene: FAP patients positive for CHRPE tend to have mutations downstream of exon 9, up to codon 1445 in exon 15; mutations in exon 9 have variable expression of CHRPE, possibly in relation to the type and location of the mutation in the exon, which is subjected to alternative splicing, patients with mutations upstream of exon 9 are CHRPE negative (40, 41, Valanzano et al., manuscript submitted). It has been hypothesised that CHRPE expression and severity of extracolorectal disease could be related to the preservation of b-catenin binding sites in the protein product encoded by the mutated allele (41). While there appears to be a correlation between APC mutation site and clinico-pathologic expression of the disease, studies of kindreds with frequent APC mutations also demonstrated a degree of phenotypic heterogeneity among patients bearing the same mutation (34,42-45). Such heterogeneity was evident even comparing patients from a single FAP-affected kindred. Following the identification of the murine homolog of the APC gene, studies on a murine model of familial adenomatous polyposis, the Min (multiple intestinal neoplasia) mouse indicated the existence on mouse chromosome 4 of at least one modifying locus, designated Mom1, responsible for partial suppression of polyp formation (46,47). Recent studies indicate that the secretory phospholipase A2 gene is a strong candidate for the Mom1 locus (48). Phospholipase A2 is a key enzyme in arachidonic acid production and is involved in cell signalling via prostaglandins and leucotrienes. Interestingly, the chromosomal band 1p35 corresponds to the position of the human phospholipase A2 gene homolog, and non-random segregation analyses of markers on chromosome 1p in FAP-affected subjects from different kindreds suggested that a human locus responsible for modifying effects on FAP maps to 1p35-36 (49,50). In accord with Knudson's "two-hit" theory (51), inactivation of both alleles of the APC gene represents an early step in the molecular pathogenesis of colorectal adenomas associated with FAP (52,53). The same mechanism takes place in sporadic gastrointestinal epithelial tumours, where somatic inactivation of both APC alleles has been consistently demonstrated (54-57). Moreover, in spite of the different histogenetic derivation, biallelic inactivation of the APC gene is also involved in the development of FAP-associated desmoids (58-60) and in the molecular pathogenesis of FAP-associated hepatoblastomas (61).
Methods of mutational analysis
The methods employed to detect mutations of the APC gene include single strand conformation polymorphism analysis (12,15, 62-64), denaturing gradient gel electrophoresis (30, 65), RNAse protection assay (55, 37), and heteroduplex analysis on polyacrylamide gels (62, 66). Other methods based on heteroduplex analysis include chemical modification and/or cleavage of mismatches, RNAse protection assay and analysis using enzymes that bind mismatched nucleotides. The selection of the APC region targeted for mutational analysis can take advantage of the observed correlations between clinical presentation and the APC mutation site. Another important aspect that should be considered in the design of diagnostic strategies concerns the mutational spectrum of the APC gene. Most mutations reported thus far introduce premature termination of protein translation. Thus, at present, various laboratories have adopted efficient methods that quickly scan large portions of coding sequence for the presence of premature stop codons (31, 67, 68). These methods use either coupled in vitro transcription/translation of PCR products (31; 68), or expression of PCR products in bacteria using an ad hoc vector that allows blue-white selection of truncated alleles (67). Another sensitive and specific diagnostic strategy based on somatic cell hybridisation, termed monoallelic mutation analysis (MAMA), has been recently proposed to detect truncating mutations. (69). MAMA may be suited for the analysis of cases in which other methods failed to detect mutations. Finally, Southern blot hybridisation may represent the technique of choice for the analysis of mutations consisting in large deletions and/or rearrangements, that would escape detection by PCR amplification of either genomic DNA or cDNA.
In any case, the above quoted techniques do not provide definitive information on the sequence of the mutations and therefore require further analysis to determine the nucleotide sequence of the mutant allele. Simplified and fast methods that do not require sequence analysis can be employed for the detection of frequent mutations. These methods, that can be performed without recurring to the use of radioisotopic tracers, include restriction enzyme digestion, when applicable, hybridisation with allele-specific probes, allele-specific PCR amplification, ligase chain reaction.
One of our laboratories at the University of Chieti uses an original strategy for the detection of three frequent APC mutations at codons 1068, 1061 e 1309 (12). This strategy is based on agarose gel heteroduplex analysis, associated with single step detection and diagnosis by multiplex allele-specific PCR.
Hereditary Non Polyposis Colorectal Cancer (HNPCC)
The prevalence of HNPCC in the general population is still uncertain. In early reports the proportion of clinically-defined HNPCC cases among all colorectal cancers ranged between 3 and 6% (70-72). However, after the adoption of the Amsterdam criteria (see below) lower estimates have been published, barely reaching 1% (73, 74). HNPCC is usually distinguished into Lynch syndrome I, which is characterised by susceptibility to colon cancer only, Lynch syndrome II, in which affected individuals show increased risk also for a variety of extracolonic epithelial-derived tumours and Muir-Torre's syndrome, where sebaceous gland tumours and skin cancers are also present.
The identifications of the genetic elements conferring an inherited susceptibility to colorectal carcinomas not associated with polyposis represented a particularly difficult challenge. Due to the relatively high frequency of colorectal cancers in the general population and to the lack of associated extracolonic or premalignant hallmarks, as in the case of FAP, the diagnosis of HNPCC is always elusive and, especially in small kindreds, true familial cases can not be easily distinguished from chance aggregation of sporadic tumours. Therefore, identification criteria were adopted in order to select for families with high incidence of colorectal tumours on the basis of common phenotypic characteristics and to provide a source of homogeneously defined kindreds for genetic analyses.
According to the so called "Amsterdam criteria", proposed by the International Collaborative Group on Hereditary Non Polyposis Colorectal Cancer, HNPCC families must fit the following requirements: a) at least three relatives must have histologically verified colorectal cancer; one of them must be a first-degree relative of the other two; familial polyposis must be excluded; b) at least two successive generations must be affected and c) one of the relatives must be below 50 years when the cancer is diagnosed (75).
Following the adoption of this working definition several HNPCC families were recruited for linkage studies to define the genomic localisation of associated genes. After intensive screening of a few hundreds genetic markers, in May 1993 positive LOD scores with DNA markers mapped to chromosome 2p was reported in two large HNPCC kindreds meeting the above mentioned criteria (76). However, it was immediately evident that genetic heterogeneity occurs in HNPCC families. In fact, a concomitant analysis of 14 smaller families led to formal exclusion of linkage to 2p for three of them, whereas the remaining 11 displayed varying degrees of positive and negative LOD scores (77). The occurrence of genetic heterogeneity was confirmed in November 1993 by the demonstration of linkage to chromosome 3p in three Swedish HNPCC families (78). Following these findings, the majority of HNPCC families were found to be associated with either 2p or 3p (79). Yet, the observation that some families were apparently unlinked to either loci suggested the existence of additional HNPCC genes.
The first indication on the nature of HNPCC genes derived from the observation that in HNPCC patients the great majority of tumours display a tendency to hypermutability of microsatellite DNA repeats (77). Microsatellites are short interspersed DNA sequences consisting of the variable repetition of mono-, di-, tri- and tetranucleotide motifs, the most frequent of which in eukaryotes is the (CA)n dinucleotide (80). The high degree of variability of the majority of microsatellite repeats makes them particularly attractive for linkage studies. In addition, microsatellites can be utilised to monitor the inactivation of tumour suppressor genes. This is accomplished through loss of heterozygosity (LOH) studies, that may reveal the occurrence of DNA losses in tumour cells, by the comparison of the allelic patterns of polymorphic markers in the constitutional and tumour DNA of the same individuals.
Most inherited cancer genes behave as tumour suppressor genes. In fact, tumours from patients carrying predisposing mutations often display the loss of the constitutionally wild-type allele (51). Therefore, following the mapping of the first HNPCC gene to chromosome 2p, the occurrence of LOH affecting this region was investigated in tumours from linked families, using the same microsatellite markers that had been employed for linkage studies. Contrary to the expectations, allelic losses were detected very rarely. However, an unpredicted finding was observed. This consisted of shifts in the electrophoretic mobility of (CA)n dinucleotide fragments, in tumour as compared to normal DNA, that were due to the increase or decrease in the number of repeats (77). Such alterations were referred as microsatellite instability.
Prior to the above illustrated findings, the occurrence of microsatellite instability had been observed in yeast DNA, where it was associated with mutations in genes whose products control the fidelity of DNA replication by correcting base pair mismatches (81). Therefore, it was hypothesised that the genomic instability detectable in HNPCC tumours was due to mutations in the human homologs of these genes (termed MMR from mismatch repair) and that such mutations introduced in tumour cells a high rate of replication errors (RERs). Moreover, it was also speculated that these mutations represented the causative event of HNPCC itself.
The formal confirmation of this hypothesis was provided by the finding that one human homolog of the bacterial gene MutS, termed hMSH2 by virtue of its sequence similarity to the yeast MSH2 gene, maps to 2p, within the same interval of one of the two HNPCC genes identified through linkage analyses (82, 83). In E. coli the MutS protein, which is part of the MutHLS excision repair pathway (84), binds mismatched nucleotides, promoting the action of the other components of the repair system (85). The finding that hMSH2 was mutated at germline level in 2p-linked HNPCC families provided definitive demonstration of its association with the syndrome (83).
In subsequent analyses, another human MMR gene was found to map to 3p, on the same region of the second identified HNPCC locus, and to be mutated in HNPCC families (86, 87). This gene, called hMLH1, displays high homology with the bacterial MutL and the yeast MLH1 genes. In E. coli the MutL gene facilitates the interaction between MutS bound to the mismatch and another protein of the repair complex, MutH, that is responsible of the excision of the newly synthesised unmethylated DNA strand (88, 89). Eventually, two additional human MutL homologs, hPMS1 and hPMS2, which map to chromosomes 2q and 7p, respectively, were isolated by screening a database of human genes identified by the expressed sequence tag (EST) method (90). Both genes were found to be mutated in the germline of HNPCC patients.
More recently, a fifth human MMR gene, named GTBP (GT binding protein) has been cloned (91, 92). GTBP maps within 0.5 megabases from hMSH2 and its product forms functional complexes with the product of hMSH2. Mutations of GTBP cause the instability of mononucleotide repeats, at variance than mutations in the other MMR genes, that are preferentially associated with (CA)n repeat instability (93). In addition, no germline mutations of GTBP have been observed in HNPCC families so far tested (93, 94, Pensotti et al., manuscript submitted).
The mechanism through with germline mutations in MMR genes confer susceptibility to cancer development is not yet fully understood. It has been postulated that alterations in a particular class of genetic elements, which include the MMR genes (mutator genes), controlling the synthesis and replications of DNA, would lead to an increase of the mutation rate in neoplastic cells (95). In this manner, the cell proceeds towards the acquisition of a fully malignant phenotype through the accumulation of mutations in cancer-related genes. However, the inactivation of both alleles of MMR genes was found to be necessary for the manifestation of the RER phenotype (96, 97). This is in keeping with the observation that tumour cell lines carrying homozygous mutations of MMR genes are deficient in repairing DNA mismatches in vitro, whereas heterozygously mutated lymphoblastoid cell lines maintain a significant repair capacity, although reduced in comparison to wild-type controls (98). On the basis of these observations, one can speculate that the cells of individuals with constitutional mutations of MMR genes are more likely to develop a "mutator phenotype" and, therefore, are more prone to neoplastic transformation, than the cells of wild-type individuals, since only one MMR allele must be inactivated for them to become genetically unstable. An apparent contradiction to this scenario is provided by the observation of microsatellite instability in heterozygous lymphoid cells from 3 HNPCC patients (99). One possible interpretation for these findings is that specific mutations may act through a dominant negative effect, which has been demonstrated for a hPMS2 truncating mutation (100).
A direct demonstration of the link between inactivation of MMR genes and tumourigenesis derive from the studies on MSH2 and PMS2 knock-out mice. In fact, although perfectly viable, these mice show a much higher tendency to develop cancer at early age than wild-type and heterozygous controls (100-102).
Few clues as to the possible nature of target loci that may trigger the mutator properties of altered MMR genes have been provided so far. In principle, the genes involved in the development of HNPCC and sporadic tumours might not be different. In fact, a colorectal carcinoma from a carrier of a constitutional hMSH2 mutation was found to harbour 6 and 4 different mutations in the APC and TP53 genes, respectively (103) and both genes are known to be mutated in a large fraction of sporadic colorectal carcinomas (104). However, whereas mutations in the 10-bp polyadenine tract within the coding region of the transforming growth factor ß type II receptor gene were detected in the vast majority of RER+ colorectal cancers, these same mutations do not occur in RER- tumours (105-107). These studies not only suggest that MMR genes may mediate tumour development through specific genetic alterations, but also provide a possible basis to explain the tumour spectrum observed in association with HNPCC. In fact, in addition to high predisposition to colorectal cancer, HNPCC patients show an increased risk for different epithelial tumours, including carcinomas of the endometrium, stomach, urinary tract, pancreas, small bowel and ovary (108). Since transforming growth factor ß is a growth inhibitor of multiple epithelial cell types (109), it is possible to speculate that the targeted inactivation of one of its receptors may selectively lead to the onset of carcinomas, but not of other tumour types.
Finally, the recent demonstration of a possible connection between the mismatch repair system and cell cycle checkpoints (110) suggests that mutations in MMR genes may elicit their tumourigenic action not only by increasing cellular mutation rate, but also through a deregulation of cell proliferation.
To date approximately 100 mutations of hMSH2 and hMLH1 have been reported (Tables 1 and 2). In contrast, mutations of hPMS1 and hPMS2 have been found only in 1 and 2 unrelated families, respectively (90, 111).
The great majority of hMSH2 and hMLH1 mutations are "unique", in that each of them has been detected only in a single family. However, some recurrent mutations have been reported, the most frequent of which is an A-T transvertion at nucleotide +3 of hMSH2 intron 5, leading to the in frame deletion of exon 5. This mutation has been found in 4 North-American, 4 British and 2 Italian HNPCC families (94, 112, Pensotti et al., manuscript submitted). In addition, two founding mutations in hMLH1, one consisting in a 3.5-kilobase genomic deletion encompassing exon 16 and another destroying the splice acceptor site of exon 6 have been observed in more than 60% of Finnish HNPCC families (113).
Mutations of hMSH2 and hMLH1 are almost uniformly distributed along the entire coding sequences. The mutational spectrum includes different types of alterations, with some noticeable difference between the two genes. In hMSH2 mutations leading to the premature termination of translation product, i.e. nonsense and frameshifts, account for approximately 40% of the total, whereas only 20% of mutations in hMLH1 fall into this category. Conversely, missense mutations are frequent in hMLH1 (approximately 30% of the total), but relatively rare in hMSH2 (less than 10%). However, the significance of the latter mutations remains undefined. In fact, in the absence of functional tests measuring the effects of aminoacid substitutions on protein activity, it is not possible to rule out that at least some of the underlying nucleotide changes represent simple DNA polymorphisms. Even the segregation of a missense mutation with the disease within a family can not be taken as a definitive proof of its causative role, since it may simply reflect the occurrence of a linkage disequilibrium with the real predisposing mutation.
Interestingly, a large fraction, approximately 40%, of both hMSH2 and hMLH1 mutations have been found to affect mRNA splicing sites or to consist in the loss of one or more exons, identifiable at cDNA level. However, it has to be remarked that in several instances such alterations have been not completely characterised, since either the effect of mutations at splicing sites on mRNA integrity could not be demonstrated or the genomic alterations underlying cDNA changes remained unidentified.
In the studies published so far, which examined populations of different origin, the mutation frequency ranged from 21% to 31% for hMSH2 (94, 114) and from 21% to 35% for hMLH1 (94, 115-117). As to the latter gene, the results obtained are in keeping with linkage analyses, estimating that approximately 30% of HNPCC families are associated with hMLH1 (78, 79). On the contrary, the proportion of families carrying germline hMSH2 mutations was lower than that (~ 50-60%) predicted by linkage studies (77, 79). It is possible that such discrepancy depends, at least in part, on inter-ethnic variability among the populations analysed for linkage studies, prevalently of North-European (Anglo-Saxon) ancestry, and those screened for mutations. However, technical limitations of the methods adopted for mutation analysis should also be taken into account. In fact, most studies employed single "indirect" screening methods, such as SSCP (single strand conformation polymorphism) analysis, IVSP (in vitro synthesised protein) assay and DGGE (denaturing gradient gel electrophoresis), whose sensitivity does not reach 100%. On the other hand, it has been demonstrated that the simultaneous use of different techniques (94) or the adoption of more complex methodologies (69, 118) increases the rate of mutation detection in MMR genes. In addition, it is possible that at least a fraction of families screened for mutations were not actually linked to MMR genes. In fact, in a recent study approximately one third of HNPCC families meeting the Amsterdam criteria were found to be negative for the presence of the RER phenotype in tumours (119). Therefore, in these families the clustering of colorectal carcinomas could have been due to chance aggregation or to the effect of genes different from those so far identified.
The latter finding indicates that the ascertainment of the RER status can represent an useful indicator for selection of HNPCC families to be screened for mutations in MMR genes. However, microsatellite instability can be detected not only in HNPCC-associated tumours, but also in 10 to 15% of apparently sporadic colorectal cases (77, 119-122). It is conceivable that at least a subset of these tumours occur in individuals in which the occurrence of a genetic predisposition cannot be inferred from family history, either because they represent de novo germline mutations, or because they belong to non informative families. This hypothesis has been recently confirmed by the report of germline MMR genes mutations in young patients (under 35 years of age) with RER+ colorectal carcinomas, but without family history of cancer (123). Nevertheless, the lack of significant association between microsatellite instability and family history of colorectal cancer, reported by a few groups (122, 124), indicates that the RER phenotype can not be considered as evidence for an inherited syndrome. In fact, it is likely that in the majority of sporadic RER+ colorectal carcinomas DNA instability is due to two somatic events that inactivate both alleles of an MMR gene (96, 97).
Form what it has been illustrated so far, it is evident that the search for mutations in HNPCC individuals is a rather difficult task. The main factors of complication are represented by: 1) the genetic heterogeneity of the disease; 2) the lack of mutational hot-spots; 3) the involvement of mutations of different types and 4) the lack of well established clinical criteria that can provide non ambiguous identification of gene carriers. As a consequence, molecular analyses are time consuming and expensive and mass screenings of colorectal cancer patients result at present unfeasible. In addition, genotype-phenotype correlation studies, that in principle could provide useful indications to address molecular investigations, are still in a preliminary phase. A risk analysis in 19 families with mutations at hMSH2 or hMLH1 showed no between-loci heterogeneity of age-specific risks for both colorectal and endometrial cancer, whereas only hMSH2 mutation carriers had a significantly increased relative risk of cancer of the urinary tract, stomach and ovaries (125).
Finally, it has to be noted that germline alterations of MMR genes are not restricted to HNPCC. In fact, a hPMS2 mutation was observed in a patient with Turcot's syndrome (111), a disease that is usually associated with mutations in the APC gene.
The discovery of the genetic elements associated with FAP and HNPCC has contributed significantly to elucidate the molecular mechanisms of tumour development in these syndromes. In addition, it allowed to demonstrate the involvement of one such element, the APC gene, in most colorectal carcinomas, whereas the exact role of MMR genes in sporadic cases still remains to be determined.
Mutational screenings of these genes have been applied to the presymptomatic diagnosis in at-risk individuals and, as for HNPCC, also to the identification of colorectal cancer cases due to inherited susceptibility (see accompanying paper by Lindblom and Björ), although this task is complicated by the heterogeneity of associated genetic lesions.
In perspective, it is possible that the genetic acquisitions will be used to modify the course of the disease trough chemoprevention or gene therapy.
Clearly, a consistent amount of research work is still to be accomplished. The genetic determinants of hereditary "hamartomatous" polyposes are still to be identified, whereas the contribution of hereditary tumour associated genes, such as BRCA1, that are known to confer an increased risk for colorectal cancers (126), to the total incidence of these tumours remains to be determined.
The occurrence of inter- and intrafamilial phenotypic heterogeneity both in FAP and HNPCC families (125, 127) suggests that, in addition to environmental factors, some genetic elements, the so called "modifying genes" such as Mom1 (47), play a role in modulating the effects of major predisposing loci. Moreover, it has been recently reported that the majority of colorectal cancer families not fitting the Amsterdam criteria are linked neither to known MMR genes, nor to genes, such as DCC, APC and TP53 that are mutated in sporadic tumours, indicating that still unidentified loci are likely to be responsible for a large fraction of familial aggregation of colorectal carcinomas with lower penetrance and later disease onset (128). It is not difficult to predict that the hunt for new genes, directly on indirectly linked to susceptibility to colon cancer will represent one of the most fascinating challenge of cancer genetics for the next future.
The work of the authors is partially supported by grants from the Italian Association for Cancer Research (AIRC), Special Project "Hereditary Tumours of the Colon".
1. American Cancer Society. Cancer Facts and Figures. 1995.
2. Macklin MT. Inheritance of cancer of the stomach and large intestine in man. J. Natl. Cancer Inst. 1960; 24: 551-571.
3. Goldgar DE et al. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J. Natl. Cancer Inst. 1994; 86: 1600-1608.
4. Rustgi AK. Hereditary gastrointestinal polyposis and nonpolyposis syndromes. N. Engl. J. Med. 1994; 331: 1694-1702.
5. Utsunomiya J. Pathology, genetics, and management of hereditary gastrointestinal polyposes. In Lynch HT, Hirayama T (eds): Genetic Epidemiology of Cancer. Boca Raton, FL: CRC Press, 1989; pp 219-149.
6. Rhodes M and Bradburn DM. Overview of screening and management of familial adenomatous polyposis. 1992; Gut 33: 125-131.
7. Bertario L et al. Cause of death and post-surgical survival in familial adenomatous polyposis: results from the Italian registry. Semin. Surg. Oncol. 1994; 10: 225-234. 8. 8. Leppert M et al. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N. Engl. J. Med. 1990; 322: 904-908.
9. Lynch, HT et al. Hereditary flat adenoma syndrome: a variant of familial adenomatous polyposis? Dis. Colon Rect. 1992; 35: 411-421.
10. Spirio L et al. Linkage of a variant or attenuated form of adenomatous polyposis coli to the adenomatous polyposis coli (APC) locus. Am. J. Hum. Genet. 1992; 51: 92-100.
11. Hodgson SV et al. Genetic heterogeneity of congenital hypertrophy of the retinal pigment epithelium (CHRPE) in families with familial adenomatous polyposis. J. Med. Genet. 1994; 31:55-58.
12. Groden J et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66: 589-600.
13. Joslyn G et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell 1991; 66: 601-613.
14. Kinzler KW et al. Identification of FAP locus genes from chromosome 5q21. Science 1991; 253: 661-665.
15. Nishisho I et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991; 253: 665-669.
16. Herrera L et al. Gardner syndrome in a man with an interstitial deletion of 5q. Am. J. Med. Genet. 1985; 25: 473-476.
17. Bodmer WF et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987; 328: 614-616.
18. Leppert M et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science 1987; 238: 1411-1413.
19. Horii A et al. Multiple forms of the APC gene transcripts and their tissue-specific expression. Hum. Mol. Genet. 1993; 2: 283-287.
20. Thliveris A et al. Demonstration of promoter activity and alternative splicing in the region 5' to exon 1 of the APC gene. Cancer Res. 1994; 54: 2991-2995 .
21. Su L et al. Association between wild type and mutant APC gene products. Cancer Res. 1993; 53: 2728-2731.
22. Rubinfeld B et al. Association of the APC gene product with b-catenin. Science, 1993; 262: 1731-1737.
23. Hinck L. b-catenin: a common target for the regulation of cell adhesion by Wnt-1 and Src signaling pathways. TIBS 1994; 19: 538-542.
24. Munemitsu S et al. Regulation of intracellular b-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl. Acad. Sci. USA 1995; 92: 3046-3050.
25. Rubinfeld B et al. The APC protein and E-cadherin form similar but independent complexes with a-catenin, b-catenin, and plakoglobin. J. Biol. Chem. 1995; 270: 5549-5555.
26. Smith KJ et al. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994; 54: 3672-3675.
27. Munemitsu S et a. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994; 54:3676-3681.
28. Smith KJ et al. The APC gene product in normal and tumor cells. Proc. Natl. Acad. Sci. USA 1993; 90: 2846-2850.
29. Nagase H and Nakamura Y Mutations of the APC (adenomatous polyposis coli) gene. Hum. Mutat. 1993; 2:435-434.
30. Olschwang S et al. Germ-line mutations in the first 14 exons of the adenomatous polyposis coli (APC) gene. Am. J. Hum. Genet. 1993; 52: 273-279.
31. Powell SM et al. Molecular diagnosis of familial adenomatous polyposis. N. Engl. J. Med. 1993; 329:1982-1987.
32. Mandl M et al. Frequency of common and novel inactivating APC mutations in 202 families with familial adenomatous polyposis. Hum. Mol. Genet. 1994; 3: 181-184.
33. Smith-Ravin J et al. APC mutations associated with late onset of familial adenomatous polyposis. J. Med. Genet. 1994; 31: 888-890.
34. Cama A et al. Multiplex PCR analysis and genotype-phenotype correlations of frequent APC mutations. Hum. Mutat. 1995; 5 : 144-150.
35. Nakamura Y. The role of the adenomatous polyposis coli (APC) gene in human cancers. Adv. Cancer Res. 1993; 62: 65-87.
36. Spirio L et al. Alleles of the APC gene: an attenuated form of familial polyposis. Cell 1993; 75: 951-957.
37. Nagase H et al. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res. 1992; 52: 4055-4057.
38. Caspari R et al. Familial adenomatous polyposis: mutation at codon 1309 and early onset of colorectal cancer. Lancet 1994; 343: 629-632.
39. Caspari R et al. Familial adenomatous polyposis: desmoid tumors and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum. Mol. Genet. 1995;4: 337-340.
40. Olschwang S et al. Restriction of ocular fundus lesions to a specific subgroup of APC mutations in adenomatous polyposis coli patients. Cell 1993;75: 959-968.
41. Wallis YL et al. Genotype-phenotype correlation between position of constitutional APC gene mutation and CHRPE expression in familial adenomatous polyposis. Hum. Genet. 1994; 94: 543-548.
42. Cama A et al. A novel deletion in exon 15 of the adenomatous polyposis coli gene in an Italian kindred. Hum. Mutat. 1994; 3: 301-304.
43. Cama A et al. A novel mutation at the splice junction of exon 9 of the APC gene in familial adenomatous polyposis. Hum. Mutat. 1994; 3:305-308.
44. Giardiello F et al. Phenotypic variability of familial adenomatous polyposis in 11 unrelated families with identical APC gene mutation. Gastroenterology 1994; 106: 1542-1547.
45. Nugent KP et al. Phenotypic expression in familial adenomatous polyposis: partial prediction by mutation analysis. Gut 1994; 35: 1622-1623.
46. Su LK et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992; 256: 668-670.
47. Dietrich WF et al. Genetic identification of Mom1 a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 1993; 75: 631-639.
48. MacPhee M et al. The secretory phospholipase A2 gene is a candidate for the Mom1 locus a major modifier of APC (Min)-induced intestinal neoplasia. Cell 1995; 81: 957-966.
49. Johnson LK et al.Localization and evolution of two human phospholipase A2 genes and two related genetic elements. In: Wong P.Y.K. Dennis E.A. eds. Phospholipase A2. New York: Plenum Press 1990.
50. Tomlison IPM et al. A modifying locus for familial adenomatous polyposis may be present on chromosome 1p35-p36. J. Med. Genet. 1996; 33:268-273.
51. Knudson AG: Hereditary cancer oncogene and antioncogene. Cancer Res.1985; 45: 1437-1443.
52. Ichii S et al. Inactivation of both APC alleles in an early stage of colon adenomas in a patient with familial adenomatous polyposis (FAP). Hum. Mol. Genet. 1992; 1: 387-390.
53. Ichii S et al. Detailed analysis of genetic alterations in colorectal tumors from patients with and without familial adenomatous polyposis (FAP). Oncogene 1993; 8: 2399-2405
54. Horii A et al. The APC gene responsible for familial adenomatous polyposis is mutated in human gastric cancer. Cancer Res. 1992; 52: 3231-3233.
55. Miyoshi Y et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl. Acad. Sci. U S A 1992; 89: 4452-4456. 56. Powell SM et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235-237.
57. Miyaki M et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res 54: 3011-3020 1994.
58. Miyaki M et al. Coexistence of somatic and germ-line mutations of APC gene in desmoid tumors from patients with familial adenomatous polyposis. Cancer Res. 1993; 53: 5079-5082.
59.Sen-Gupta S et al. Somatic mutation of APC gene in desmoid tumour in familial adenomatous polyposis. Lancet 1993;342: 552-553.
60. Cama A et al. Novel mutations and inactivation of both alleles of the APC gene in desmoid tumors. Hum. Mol. Genet. 1995;4: 1979-1981.
61. Kurahashi H et al. Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res. 1995; 55: 5007-5011.
62. Cottrell S et al. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992; 340: 626-630.
63. Groden J et al. Mutational analysis of patients with adenomatous polyposis: identical inactivating mutations in unrelated individuals. Am. J. Hum. Genet. 1993; 52: 263-72.
64. Varesco L et al. Identification of APC gene mutations in Italian adenomatous polyposis coli patients by PCR-SSCP analysis. Am. J. Hum. Genet. 1993; 52: 280-285.
65. Fodde R et al. Eight novel inactivating germ line mutations at the APC gene identified by denaturing gradient gel electrophoresis. Genomics 1992; 13: 1162-1168.
66. Friedl W et al. Single-step screening method for the most common mutations in familial adenomatous polyposis. Hum. Mol. Genet. 1993; 2: 1481-1482.
67. Varesco L et al. Rapid screening method to detect nonsense and frameshift mutations: identification of disease-causing APC alleles. Cancer Res. 1993; 53: 5581-5584.
68. van der Luijt R et al. Rapid detection of translation-terminating mutations at the adenomatous polyposis coli (APC) gene by direct protein truncation test. Genomics 1994; 20:1-4.
69. Papadopoulos N et al. Monoallelic mutation analysis (MAMA) for identifying germline mutations Nature Genet. 1995; 11: 99-102.
70. Lynch HT. Frequency of hereditary non-polyposis colorectal carcinoma (Lynch syndromes I and II). Gastroenterology 1986; 90: 486-496.
71. Ponz de Leon M et al. Incidence and familial occurrence of colorectal cancer and polyps in a health-care district of Northern Italy. Cancer 1987; 60: 2848-2859.
72. Mecklin J-P. Frequency of hereditary colorectal cancer. Gastroenterology 1987; 93: 1021-1025.
73. Kee F and Collins BJ. Families at risk of colorectal cancer: who are they?. Gut 1992; 33: 787-790.
74. Aaltonen LA et al. A novel approach to estimate the proportion of Hereditary non-polyposis colorectal cancer of total colorectal cancer burden. Cancer Detect. Prev. 1994; 18: 57-63.
75. Vasen HFA et al. Hereditary non-polyposis colorectal cancer. Lancet 1991; 338: 877.
76. Peltomäki P et al. Genetic mapping of a locus predisposing to human colorectal cancer. Science 1993; 260: 810-812.
77. Aaltonen L et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260: 812-816.
78. Lindblom A et al. Genetic mapping of a second locus predisponing to hereditary nonpolyposis colon cancer. Nature Genet. 1993; 5: 279-282.
79. Nyström-Lahti M et al. Mismatch repair genes on chromosomes 2p and 3p account for a major share of hereditary nonpolyposis colorectal cancer families evaluable by linkage. Am. J. Hum. Genet. 1994; 55: 659-665.
80. Gyapay G et al. The 1993-94 Genethon human genetic linkage map. Nature Genetics 1993; 7: 246-339.
81. Strand M et al. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993; 365: 274-276.
82. Fishel R et al. The human mutator gene homolg MSH2 and its association with hereditary non polyposis colon cancer. Cell 1933; 75: 1027-1038.
83. Leach FS et al.Mutations of a mutS homolog in hereditary nonpolyposic colorectal cancer. Cell 1993; 75: 1215-1225.
84. Modrich P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991; 25: 229-253.
85. Su S.S. and Modrich P. Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc. Natl. Acad. Sci. USA 1986; 83: 5057-61.
86. Bronner CE et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non polyposis colon cancer. Nature 1994; 368: 258-261.
87. Papadopoulos N et al. Mutation of a mutL homolog in hereditary colon cancer. Science 1994; 263: 1625-1629
88. Welsh KM et al. Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem. 1987; 261: 15624-15629.
89.Grilley M et al. Isolation and characterization of the Escherichia coli mutL gene product. J. Biol. Chem. 1989; 264: 1000-1004.
90. Nicolaides NC et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994; 371: 75-80.
91. Drummond JT et al. Isolation of an hMSH2 p160 heterodimer that restore DNA mismatch repair to tumor cells. Science 1995; 268: 1909-1912.
92. Palombo F et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995; 268: 1912-1914.
93. Papadopoulos N et al. Mutations of GTBP in genetically instable cells. Science 1995, 268: 1915-1917.
94. Liu B et al. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nature Med. 1996; 2: 169-174.
95. Loeb LA. Microsatellite instability: Marker of a mutator phenotype in cancer. Cancer Res. 1994; 54: 5059-5063.
96. Hemminki A et al. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nature Genet. 1994, 8: 405-410.
97. Liu B et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nature Genet. 1995; 9: 48-55.
98. Parsons R et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 1993; 75: 1227-1236.
99. Parsons R et al. Mismatch repair deficiency in phenotypically normal human cells. Science 1995; 268: 738-740.
100. Reitmair AH et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nature Genet. 1995; 11: 64-70.
101. Baker SM et al. Male mice defective in the mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell 1995; 82: 309-319.
102. de Wind N et al. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995; 82: 321-330.
103. Lazar V et al. Accumulation of multiple mutations in tumour suppressor genes during colorectal tumorigenesisin HNPCC patients. Hum. Mol. Genet. 1994; 3: 2257-2260.
104. Marx J. New colon cancer gene discovered. Science 1993; 260:751-752.
105. Markowitz S et al. Inactivation of the type II TGFß- receptor in colon cancer cells with microsatellite instability. Science 1995; 268: 1336-1338.
106. Parson R et al. Microsatellite instability and mutations of the transforming growth factor ß type II receptor gene in colorectal cancer. Cancer Res. 1995; 55: 5548-5550.
107. Lu S-L et al. Mutations of the transforming growth factor-ß type II receptor gene and genomic instability in hereditary nonpolyposis colorectal cancer. Biochem. Biophys. Res. Commun.1995; 216: 452-457.
108. Watson P and Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 1993; 71: 677-685.
109. Wrana JL et al. Mechanism of activation of the TGF-ß receptor. Nature 1994; 370: 341-347.
110. Hawn MT et al. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint.Cancer Res. 1995; 5: 3721-3725.
111. Hamilton SR et al. The molecular basis of Turcot's syndrome. N. Engl. J. Med. 1995, 332: 839-847.
112. Froggatt NJ et al A frequent hMSH2 mutation in hereditary non-polyposis colon cancer syndrome. Lancet 1995; 345: 727.
113. Nyström-Lahti M et al. Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nature Med. 1995; 1: 1203-1206.
114. Wijnen J et al. Seven new mutations in hMSH2, an HNPCC gene, identified by denaturing gradient gel electrophoresis. Am.J. Hum. Genet. 1995; 56: 1060-1066.
115. Han H-J et al. Genomic structure of human mismatch repair gene, hMLH1, and its mutation analysis in patients with hereditary nonpolyposis colorectal cancer (HNPCC). Hum. Mol. Genet. 1995; 4: 237-242.
116. Tannergård P et al. Mutation screening in the hMLH1 gene in Swedish hereditary nonpolyposis colon cancer families. Cancer Res. 1995; 55: 6092-6096.
117. Wijnen J et al. Majority of hMLH1 mutations responsible for hereditary nonpolyposis colorectal cancer cluster at the exonic region 15-16. Am.J.Hum.Genet. 1996; 58: 300-307.
118. Nystrom-Lahti M et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum. Mol. Genet. 1996; 5: 755-762.
119. Jass JR et al. Diagnostic use of microsatellite instability in hereditary non-polyposis colorectal cancer. Lancet 1995; 346: 1200-1201.
120. Ionov Y et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363: 558-561.
121. Thibodeau S et al. Microsatellite instability in cancer of the proximal colon. Science 1993; 260: 816-819.
122. Lothe RA et al. Genomic instability in colorectal cancer: Relationship to clinicopathological variables and family history. Cancer Res. 1993; 53: 5849-5852.
123. Liu B et al. Genetic instability occurs in the majority of young patients with colorectal cancer. Nature Med. 1995; 1: 348-352.
124. Samowitz WS et al. Microsatellite instability in human colonic cancer is not a useful clinical indicator of familial colorectal cancer. Gastroenterology 1995; 109: 1765-1771.
125. Vasen HFA et al. Cancer risk in families with hereditary non polyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 1996; ???
126. Ford D et al. Risk of cancer in BRCA1-mutation carriers. Lancet 1994; 343: 692-695.
127. Presciuttini S et al. Age of onset in familial adenomatous polyposis: heterogeneity within famlies and among APC mutations. Ann. Hum. Genet. 1994; 58: 331-342.
128. Lewis CM et al. Genetic heterogeneity and unmapped genes for colorectal cancer. Cancer Res. 1996; 56:1382-1388.
129. Liu B et al. hMSH2 mutations in hereditary nonpolyposis colorectal cancer kindreds. Cancer Res. 1996; 54: 4590-4594.
130. Kolodner RD et al. Structure of the human MSH2 locus and analysis of two Muir-Torre kindreds for msh2 mutations. Genomics 1994; 24: 516-526.
131. Mary J-L et al. Mutational analysis of the hMSH2 gene reveals a three base pair deletion in a family predisposed to colorectal cancer development. Hum. Molec. Genet. 1994; 3: 2067-2069.
132. Orth K et al. Genetic instability in human ovarian cancer cell lines. Proc. Natl. Acad. Sci. USA 1994; 91: 9495-9499.
133. B¯rresen A-L et al. Somatic mutations in the hMSH2 gene in microsatellite unstable colorectal carcinomas. Hum. Molec. Genet. 1995; 4: 2065-2072.
134. Buerstedde J-M et al. Detection of new mutations in six out of 10 Swiss HNPCC families by genomic sequencing of the hMSH2 and hMLH1 genes. J. Med. Genet. 1995; 32: 909-912.
135. Liu B et al. Genetic instability occurs in the majority of young patients with colorectal cancer. Nature Med. 1995; 1: 348-352.
136. Maliaka YK et al. CpG dinucleotides in the hMSH2 and hMLH1 genes are hotspots for HNPCC mutations. Hum. Genet. 1996; 97: 251-255.
137. Kolodner RD et al. Structure of the human MLH1 locus and analysis of a large hereditary nonpolyposis colorectal carcinoma kindred for mlh1 mutation. Cancer Res.1995; 55: 242-248.
Table 1. Germline mutations in the hMSH2 gene
EXON |
Missense |
Nonsense |
Frameshift |
Splice site/ Exon skipping |
Others |
Total |
1 |
- |
- |
- |
- |
1a |
1 |
2 |
- |
- |
- |
- |
- |
- |
3 |
- |
- |
1 |
- |
- |
1 |
4 |
- |
1 |
- |
- |
- |
1 |
5 |
- |
1 |
1 |
1+10b |
- |
13 |
6 |
1 |
- |
1 |
- |
- |
2 |
7 |
- |
1+1 |
1+1 |
1+1 |
1c |
7 |
8 |
- |
1+1 |
- |
- |
1d |
3 |
9 |
- |
- |
1 |
- |
- |
1 |
10 |
1 |
- |
1 |
- |
- |
2 |
11 |
- |
- |
- |
- |
- |
- |
12 |
1 |
2+1 |
1 |
1 |
3e |
9 |
13 |
- |
- |
- |
2 |
- |
2 |
14 |
- |
- |
1+1 |
- |
- |
2 |
15 |
- |
- |
- |
2 |
- |
2 |
16 |
- |
- |
- |
- |
- |
- |
Total |
3 (6.5%) |
9 (19.6%) |
10 (21.7%) |
18(39.1%) |
6 (13.0%) |
46 |
The table lists the mutations reported in refs. 83, 94, 103, 112, 114, 129-126 and Pensotti et al., submitted. Each entry indicates a different mutation ; the numeric values indicate how many times the same mutations has been reported
adeletion of 5'end of gene
bGTA to GTT at splice donor site
c107-bp insertion
ddeletion of exon 8 to 15
ein-frame deletion of codon 596
Table 1. Germline mutations in the hMLH1 gene
EXON |
Missense |
Nonsense |
Frameshift |
Splice site/ Exon skipping |
Others |
Total |
1 |
- |
- |
- |
- |
1 |
1 |
2 |
3a+1+1 |
1 |
- |
- |
- |
6 |
3 |
- |
- |
- |
1 |
- |
1 |
4 |
2+1 |
- |
- |
- |
- |
3 |
5 |
1 |
- |
- |
- |
- |
1 |
6 |
- |
- |
- |
1+1 |
- |
2 |
7 |
- |
- |
- |
1 |
- |
1 |
8 |
- |
- |
- |
1+1+1+1 |
4 |
|
9 |
1 |
- |
- |
1+1+1 |
- |
4 |
10 |
- |
- |
- |
1 |
- |
1 |
11 |
1 |
- |
1+1 |
- |
- |
3 |
12 |
- |
- |
- |
2 |
- |
2 |
13 |
1 |
- |
1 |
1 |
- |
3 |
14 |
1 |
- |
- |
1+1 |
- |
3 |
15 |
1 |
- |
- |
1+1 |
- |
3 |
16 |
1+1 |
- |
1+1+1 |
1 |
4b+1c |
11 |
17 |
- |
- |
- |
- |
- |
- |
18 |
- |
- |
- |
- |
- |
- |
19 |
1 |
1 |
2+1 |
- |
1d |
6 |
Total |
17 (30.9%) |
2 (3.6%) |
9 (16.4%) |
22 (40.0%) |
5 (9.1%) |
55 |
The table lists the mutations reported in refs. 86, 87,94, 111, 113 115-117 134-137, and Pensotti et al., submitted. Each entry indicates a different mutation ; the numeric values indicate how many times the same mutations has been reported
a Ser_ Phe codon 44
b in-frame deletion of one codon between 616 and 618
c 3.5-kb genomic deletion; detected in ca. 50% of Finnish HNPCC families
d extension of -COOH terminus
Figure 1. Diagrams of the APC gene and protein.The structure of the coding exons of the APC gene is reported in the top panel. The regions of the gene where mutations are associated with attenuated or severe phenotype as well as CHRPE are underlined. Some of the domains of the APC protein, involved in protein-protein interactions, are indicated in the lower panel.
Figure 2. Localization of germ line mutations of the APC gene.Brackets indicate the regions of the APC gene where germ line mutations are commonly detected. The sites of some common germ-line mutations are indicated by arrows.
Figure 3. In vitro transcription-translation of PCR products.Full-length peptides (531 amino acids) derived from the transcription translation of PCR amplified exon 15 sequences are visible in lanes 1 and 2. An additional band corresponding to a truncated peptide derived from an allele bearing a deletion at codon 1061 is detected in lane 1.